




基于分子轨道理论三个基本近似的物理模型,采用一组单电子函数——分子轨道波函数描述分子中的电子运动状态,对电子数N为偶数的分子,遵守能量最小原则和泡利不相容原理,按轨道能级由低到高顺序依次将电子配对填入各个分子轨道。
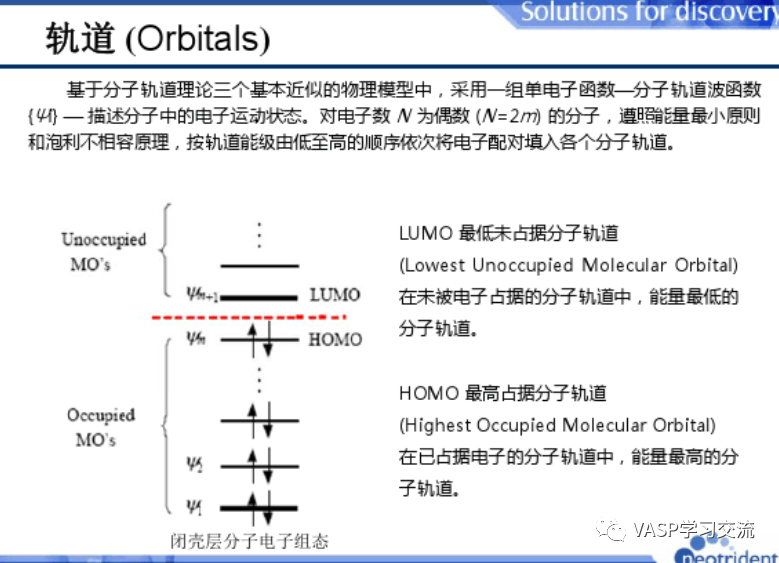
1. 预测反应性,使用MS的DMol3模块,高斯或者VASP都可以给出整个体系的分子轨道信息。对于反应来说,可以使用前线轨道理论对称守恒原理,判断发生的区域。例如:
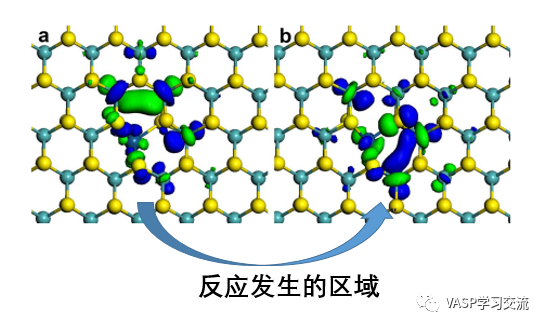
Nat. Nanotechnol. 13, 411–417 (2018).
2. 对于单个分子来说可以帮助判断电子的可能跃迁,一般来说,HOMO轨道的值越高说明越容易给电子到受体,LUMO越低,越容易接收电子;对应的HOMO-LUMO gap (其实和能带是一个概念,只是能带说的最多是周期性体系,HOMO-LUMO gap针对分子体系),通常比较低,就说明分子很容易发生化学反应。其实电子从HOMO到LUMO的跃迁是整个体系中所有可能发生的跃迁中最容易发生的,其他任何跃迁所需的能量都大于这个值,HOMO-LUMO gap越大,电子从HOMO到LUMO的跃迁的所需要吸收的光的能量越大,吸收波长也就越短,跃迁也就相对不容易发生或者发生的概率低,导致光化学活性低。
3. 在这里已经教了如何计算小分子开展一个计算了,今天就用VASP来计算N2的分子的HOMO, LUMO(一般分子的HOMO,LUMO用高斯计算最多,如果想要更精确的结果,可以使用高斯计算)
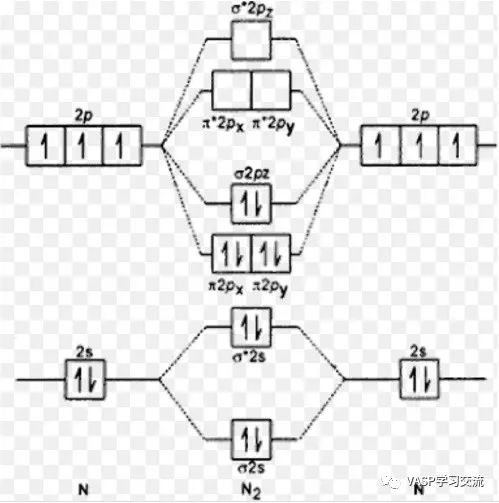
这个图给出N2分子的轨道排布图,σ2pz成键轨道对应于HOMO,π*反键轨道对应于LUMO
4. VASP计算HOMO LUMO
4.1 INCAR的准备,这里是静态计算了,需要WAVECAR,所以INCAR参数需要做以下修改:
NSW=0
LWAVE=.TRUE.
ISPIN=1 #N2分子没有磁矩,所以我把自旋关了
#别的计算参数一律不变
4.2 提交作业,结束后查看OUTCAR,直接从末尾开始往上翻(键盘同时按住 Shift + G键,自动跳到末尾)
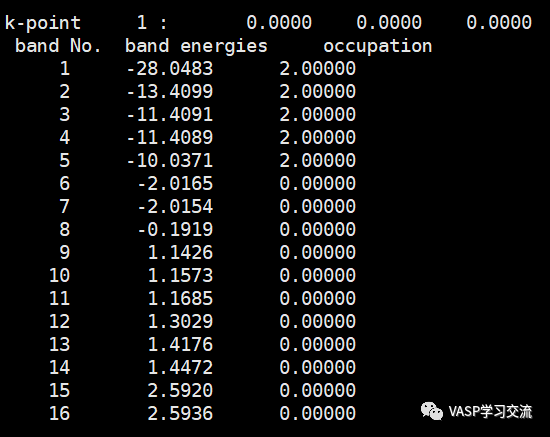
图中 band No 对应能级的序号, band energies对应于能级的位置,occupation对应于占据数,占据数一般可以为0 1 2(我这里就是2,因为我关了自旋),如果分子体系出现分数,就要注意有没有出现错误,具体的可以去大师兄科研网O2计算那一节看看,这里就不多加赘述,对于金属体系,有可能出现分数占据
结合上面的分子轨道能级图:
可以发现能带3和能带4能量相近,是能级简并的,应该对应于上图的π2px和π2py轨道,能带5对应的是图中的σ2pz轨道,也就是最高占据态HOMO,对应的能级是-10.0371 eV,因为能带5占据数是2,能带6,7就是空占据态了,而且能级能量相近,对应于上图中的π2px和π2py反键轨道,这两个轨道也是简并的,因为也就是最高未占据态LUMO,对应的能级是-2.0165 eV,再往下就是能带8了,对应于上图中的σ2pz轨道。其实我们最终看的也就是能带5 和能带6 or 7了,HOMO-LUMO gap=8.02 eV。
5 可视化HOMO LUMO
#使用万能的vaspkit,如有使用请引用一下,vaspkit前面已讲过如何配置了
往期推荐
输入vaspkit输入511——输入1——5——先得到能带5的吧,以此类推得到能带6

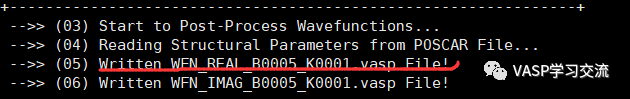
把得到的WFN_REAL_B0005_K0001.vasp文件下载下来,直接拖入到VESTA作图如下,后面想得到多个能带,按照步骤依次画就行。
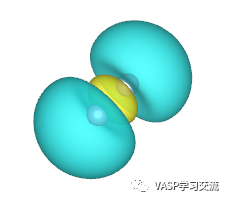
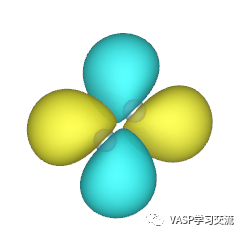
左图对应HOMO,右图对应LUMO
