




一直以来对epigenetic分析都有一些恐惧,因为本科的时候就没怎么搞懂,到现在看见chip-seq啥的就头疼。之前看刘小乐教授的课程也是一知半解。幸亏看到孟浩巍大神在知乎live上有讲这期,于是欣喜地学习之~
Epigenetic理论部分
基因组profiling
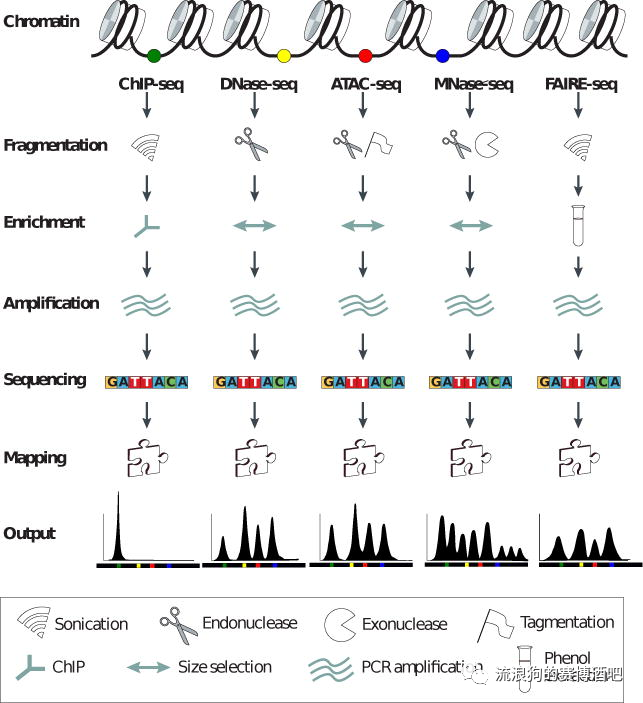 图片来源(Meyer CA, Liu XS. Nat Rev Genet. 2014;15(11):709-721)
图片来源(Meyer CA, Liu XS. Nat Rev Genet. 2014;15(11):709-721)
核小体:
核小体是染色质的基本结构单位,由DNA和H1、H2A、H2B、H3和H4等5种组蛋白(histone,H)构成。两分子的H2A、H2B、H3和H4形成一个组蛋白八聚体,约200 bp的DNA分子盘绕在组蛋白八聚体构成的核心结构外面1.75圈形成了一个核小体的核心颗粒(core particle)。核小体的核心颗粒再由DNA和组蛋白H1共同构成的连接区连接起来形成串珠状的染色质细丝。
chip-seq:
打断:超声破碎 富集:抗体结合。也就是,已知某种蛋白与DNA的特定区域结合,那么用这种蛋白抗体就可以将打断后的片段中含有目标DNA的部分进行富集了。 纯化:用PCR等实验方法对获取的目标片段建库 测序:对目标片段测序 map:将测得的序列mapping到参考序列中 output;又称为peak calling。
chip-seq最终得到的是与目标蛋白结合的序列在DNA上的位置信息。(组蛋白以及转录因子结合区域)
DNase-seq、ATAC-seq、MNase-seq、FAIRE-seq
目标是得到染色体DNA有哪些是打开的。也就是哪些地方能够进行比较活跃的转录,哪些地方比较不活跃。
Hi-C:一种3D基因组技术
Hi-C的实验例程

染色体在细胞内的分布是三维结构,蛋白质互相交联以稳定构象。 用限制性核酸内切酶cut形成粘性末端片段 在补平粘性末端时加入biotin标签。 进行平末端的连接 超声打碎,形成带biotin标签和不带标签的两种片段。用biotin抗体把带标签的片段pull down。 对pull down的片段进行双端测序。被测序的片段两边是来自于序列距离较远而同时在空间结构上靠近的染色体。
Hi-C的数据处理及可视化
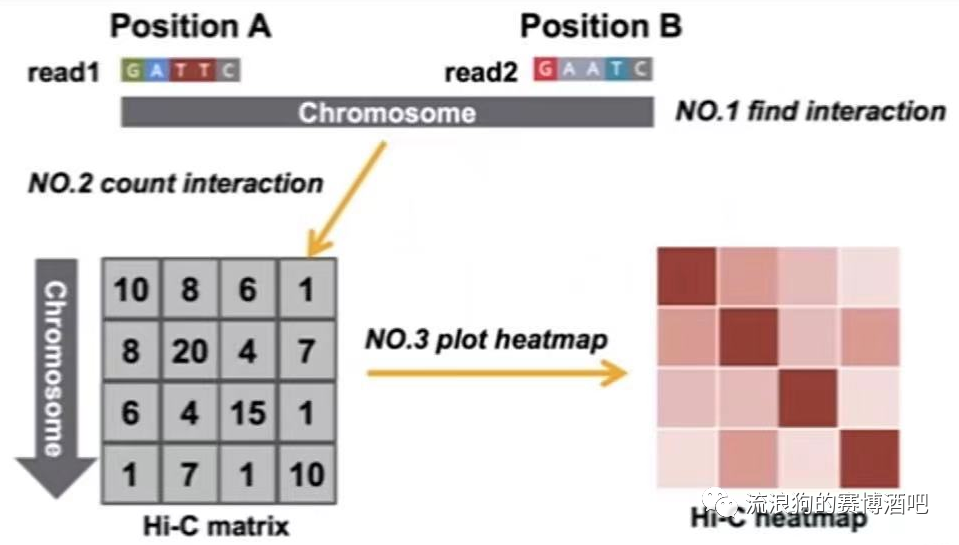 在整条染色体上寻找互作位点并且计数。形成heatmap,深色区域代表两个position互作关系较强。
在整条染色体上寻找互作位点并且计数。形成heatmap,深色区域代表两个position互作关系较强。
基因组区域的划分
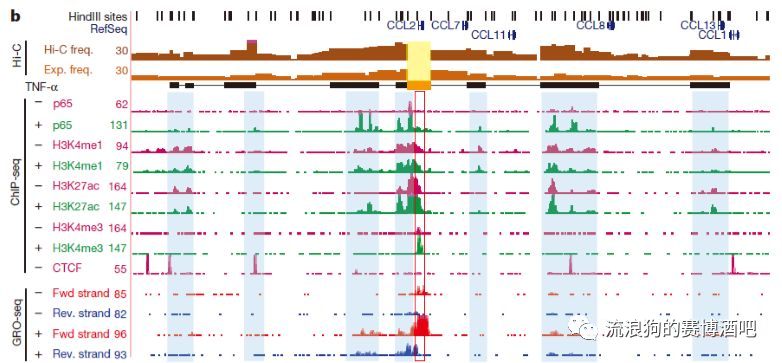 chip-seq行名含义,以H3K27ac为例:H3是组蛋白名称,K27是指27号位的氨基酸K(赖氨酸),ac指乙酰化修饰。
chip-seq行名含义,以H3K27ac为例:H3是组蛋白名称,K27是指27号位的氨基酸K(赖氨酸),ac指乙酰化修饰。
不同的组蛋白修饰会形成不同的功能。例如H3K4me3一般代表活跃信号,也就是说有这种修饰出现时,代表这个区域时转录活跃的区域。H3K9me3,代表抑制信号。而这种修饰的不同可以把染色体分为不同的区域。(强启动子、弱启动子、强增强子、转录区等)
ENCODE数据库:基因元件
可以下载相应数据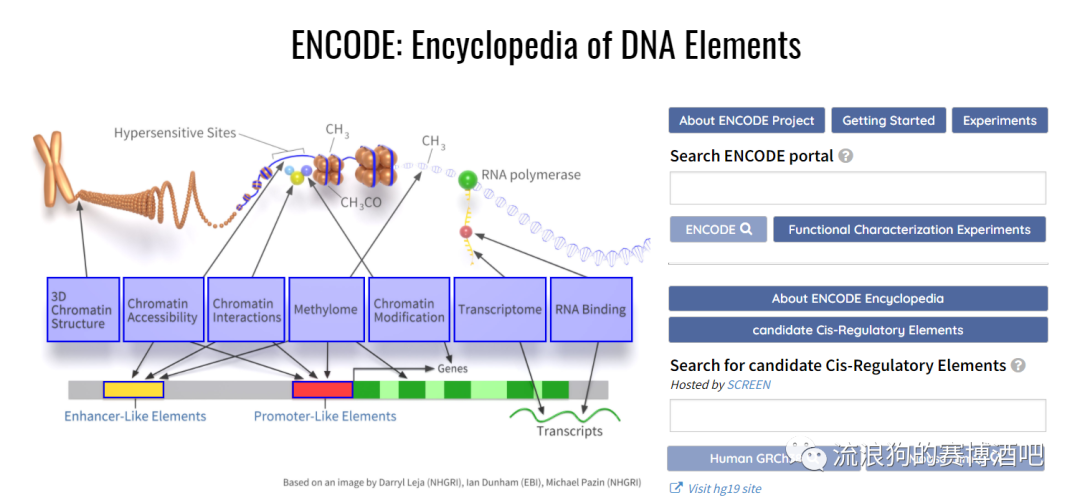
chip-seq Figure解读
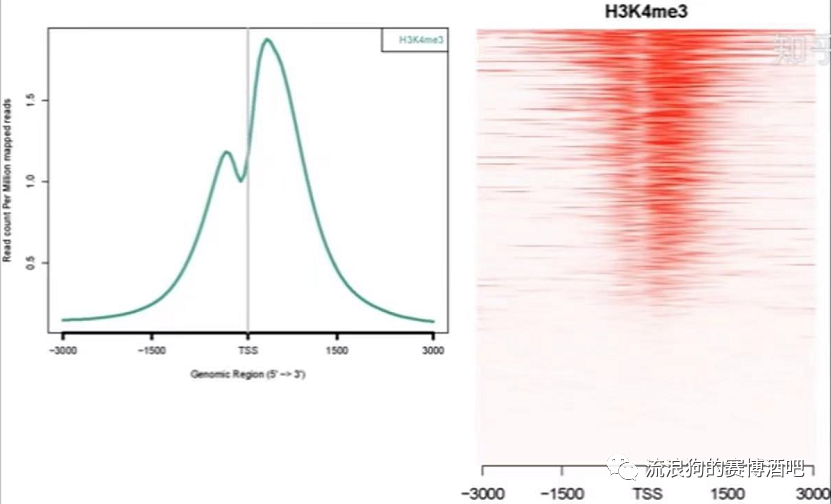
左图
横轴:TSS(转录起始位点)上下游位置
纵轴:reads count
图示了reads在转录起始位点前后的分布情况。
右图
红色深浅代表reads count,与左图其实同一个意思。
补充
其实Hi-C的数据可视化孟大佬没讲透,还有loop、TAD和compartments这些互作关系,可以参考这个知乎答案(三维基因组学研究的是什么?什么是 Hi-C 技术?为什么 Hi-C 技术这么火?- 张一柯的回答 - 知乎 https://www.zhihu.com/question/50270622/answer/1715108352)
