




BLAST 算法
课程视频:https://www.youtube.com/watch?v=g0nSH17psDc
BLAST是基于局部比对的算法,本视频对序列比对相关内容没有进行讲解。
What is blast ?
BLAST(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。BLAST程序能迅速与公开数据库进行相似性序列比较。BLAST结果中的得分是对一种对相似性的统计说明。
Types of blast

BLASTP是蛋白序列到蛋白库中的一种查询。库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
BLASTX是核酸序列到蛋白库中的一种查询。先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
BLASTN是核酸序列到核酸库中的一种查询。库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
TBLASTN是蛋白序列到核酸库中的一种查询。与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
TBLASTX是核酸序列到核酸库中的一种查询。此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
其中,最迅速的是DNA-DNA比对以及protein-protein比对。
Heuristic Search

左边有一段序列是相同的,而右边不存在相同序列。这种情况下,BLAST会选择左边而忽略右边。这样使BLAST算法变得更快速。
Algorithm
算法流程:

1. Find all possible words in query
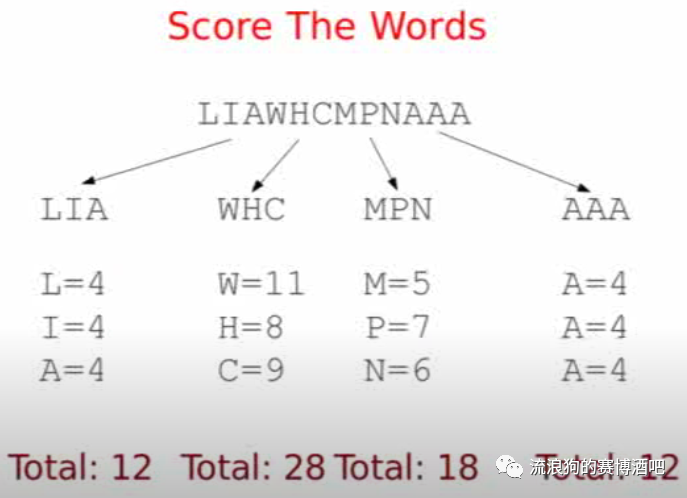
例如一段氨基酸序列为LIAWHCMPNAAA,把它分为4个k=3的k-mer,根据BLOSUM62矩阵,每个K-mer最高得分如下:
BLOSUM62 matrix
BLOSUM62打分细节可以参考:https://blog.csdn.net/weixin_43770577/article/details/104023846
得到分数后,通过指定一个threshold把得分较低的K-mer去掉,只留下WHC 和MPN。
2. Scan the database for occurrence of words

在database中寻找出现在query中的words(这一步计算机可以很快完成)
3. Score the alignment

从k-mer向两个方向延伸,如果variant在矩阵中得分为正则继续延伸(extend),如果无法延伸则跳过这些不能配对的部分(join the gaps)。
等到延伸至无法继续时,计算所有配对的序列,得到一个HSP值

根据矩阵计算所有匹配得分。另外gap open和gap extension需要另算罚分。最终得分为54分。

Analyse the score

Align and score to same sequence
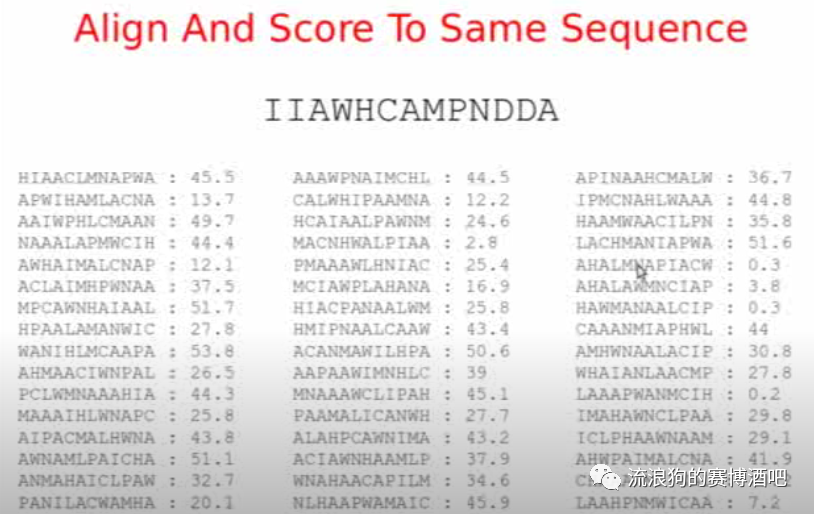
把query string的所有排列组合可能性列出,分别与database里被找到的这一条序列进行align,分别计算他们的得分。
plot score
把这些得分做一个直方图,形状如图。
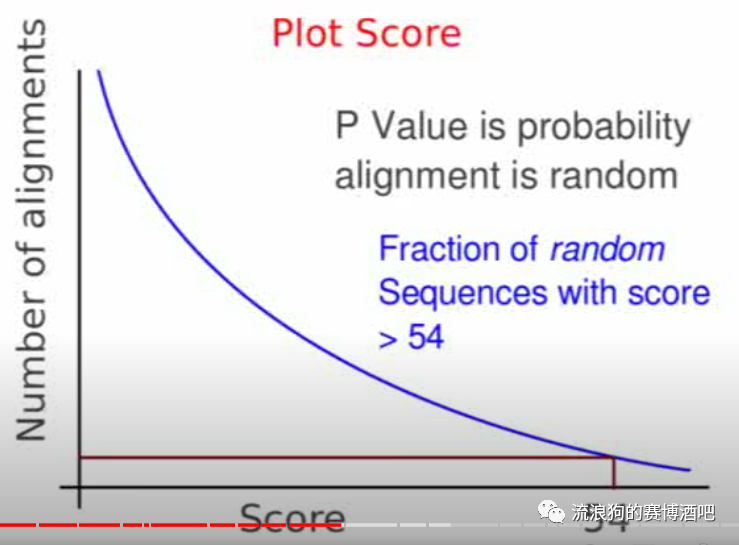
图中可以观察到:得分超过54的序列相比于随机序列占比很低,这说明随机顺序的序列不太可能得分超过54,所以在database找到的这条序列是具有显著性的。***(这里有一个问题,为什么这里仅列举组分相同的随机序列,而不是把所有氨基酸的排列组合可能性都列举出来?)***
这是根据经验得到的概率阈值。

Convert P-values to E-values

BLAST算法还有一个oversample(过采样)问题。越长的序列和database就越容易得到高分。为避免过拟合问题,需要将P值转变为E值

database长度 x query序列长度 x 概率
计算公式如下。λ和k是scaling factor,无实际意义,在输出结果处可以查看。

Convert raw score to bit score

Bit score to E value

当数据库变大,E value也会随之变化。
重要关系!!:当bit score(S')变大,E value 会降低
filtering

图中是使用TBLASTX对核酸序列进行匹配(先转换为氨基酸,再通过氨基酸匹配计算得分)图中4个氨基酸全都是proline,但是下面那行全都是C。看起来不像一个正常的核酸序列。所以BLAST设计了过滤环节,目的是去除这种低复杂度序列。
有两种方法

上面是soft filtering,忽视这段序列,不改变序列长度;
下面是hard filtering,把这段序列转换为N,并改变序列长度(以及E值)
下图为常用过滤工具

psi-BLAST
首先我们有一些已比对的氨基酸序列,如下图

根据这些序列中氨基酸的出现概率,得到一个矩阵
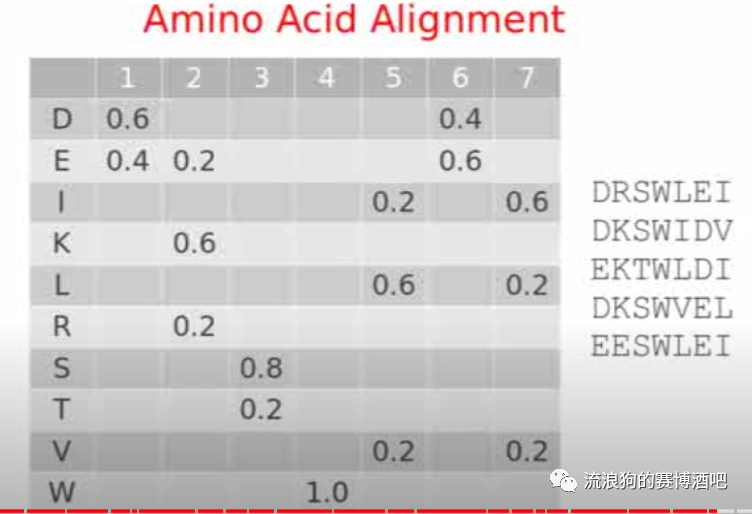
当我们得到一个新的序列的时候,就可以用这个矩阵对序列进行打分。
把分数相加后,除以序列总长度。这个分数可以帮助我们判断新序列是否与已知的比对序列有相关关系。
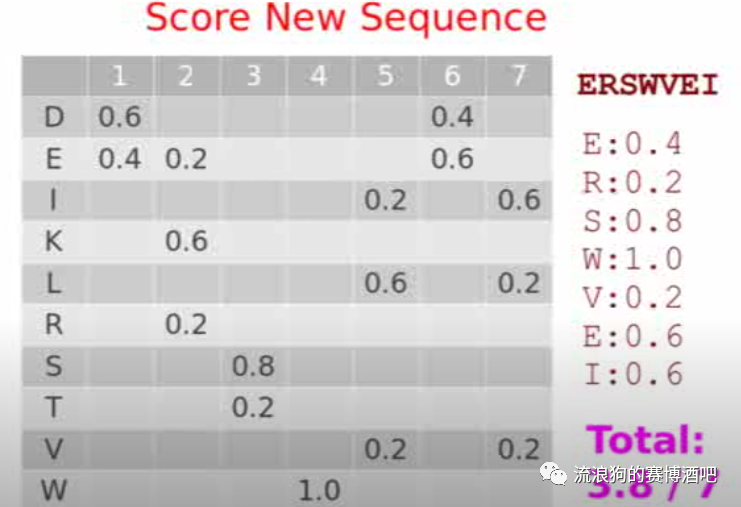

总结。

