




Spatial Single-Cell Transcriptomics Toolbox • Giotto (rubd.github.io)
https://rubd.github.io/Giotto_site/
上一次的推文Giotto—空间转录组工具箱(一)已经涉及到了如何通过单细胞数据注释空转的spot,所使用的默认方法叫PAGE(Parametric Analysis of Gene Set Enrichment),它的原理如下:首先计算spot相比其他spot所有基因,差异倍数的均值和标准差,然后计算基因集(也就是单细胞得到的sig)在spot中的差异变化的均值,根据下面的公式可得ES,该值越大越代表细胞类型与spot相关(我的理解就是ES越大越代表变化的差异值都是由基因集提供的,越能说明两者的相似性)

代码和上次一样,最重要的就是要自己定义好基因集:
load("sc_marker.Rds")sc.marker<-sc.marker%>%filter(p_val_adj<0.05)sc.marker.select<-sc.marker %>% group_by(cluster) %>% top_n(50,wt=avg_log2FC)sc.marker.select<-sc.marker.select[,c("cluster","gene")] %>% split(.$cluster) %>% lapply( "[[", 2)sig_matrix<-makeSignMatrixPAGE(names(sc.marker.select), sc.marker.select)## enrichment testsmini_visium = runSpatialEnrich(mini_visium,sign_matrix = sig_matrix,enrich_method = 'PAGE')
同样Giotto还提供了另外两种细胞富集的思路:rank和hypergeometric distribution,这里先看一下hypergeometric distribution(超几何分布)。实际上我在课上给大家讲解的MIA就是基于超几何分布的,Giotto的做法是首先根据是否有marker基因以及二分化基因表达(top 5%)将基因分成四类,然后进行超几何分布检验,以-log10(P value)作为ES,ES越大越说明高表达的差异基因越多,越代表两者的相似性。

mini_visium = runSpatialEnrich(mini_visium,sign_matrix = sig_matrix,enrich_method = 'hypergeometric')
最后是rank,它不是基于基因集的方法,而是使用单细胞所有的表达数据。首先计算某基因在特定细胞中,相较于其他细胞类型的fold change,然后根据fc值将基因排序得到秩次R1,接着计算该基因在某spot中相较于其他spot的fold change,再次排序得到R2,最后相乘取开方,得到Rg。通过公式计算RBP,RBP高的基因同时拥有单细胞中高表达以及空间位置上高表达两个特性,随后计算前100个RBP的和得到ES。

# 这里代码要稍作更改:sig_matrix_rank<-makeSignMatrixRank(scRNA@assays$RNA@counts,scRNA$celltype,ties_method = "max",gobject = mini_visium)## enrichment testsmini_visium = runSpatialEnrich(mini_visium,sign_matrix = sig_matrix_rank,enrich_method = 'rank')
接下来分别可视化一下:
cell_types = colnames(sig_matrix)plotMetaDataCellsHeatmap(gobject = mini_visium,metadata_cols = 'leiden_clus',value_cols = cell_types,spat_enr_names = 'PAGE',x_text_size = 8, y_text_size = 8)+plotMetaDataCellsHeatmap(gobject = mini_visium,metadata_cols = 'leiden_clus',value_cols = cell_types,spat_enr_names = 'hypergeometric',x_text_size = 8, y_text_size = 8)+plotMetaDataCellsHeatmap(gobject = mini_visium,metadata_cols = 'leiden_clus',value_cols = cell_types,spat_enr_names = 'rank',x_text_size = 8, y_text_size = 8)
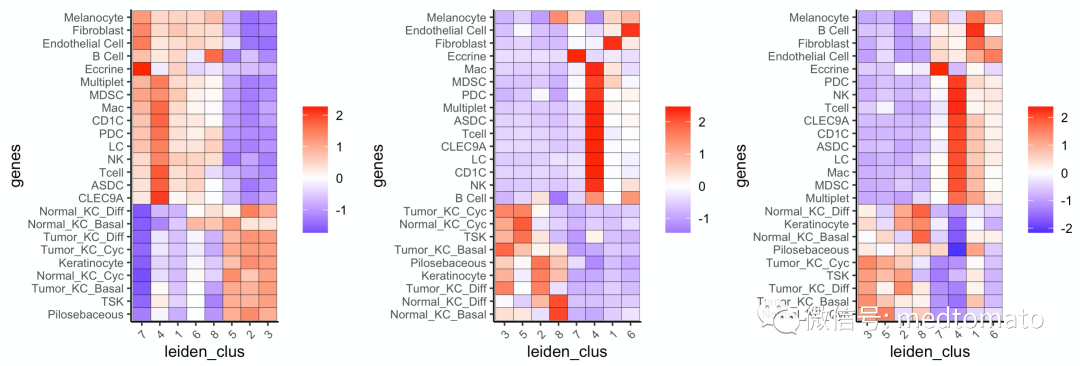
可以看到结果还是比较稳健的~
Identify spatial genes with 3 different methods:
- binSpect with kmeans binarization (default)
- binSpect with rank binarization
- silhouetteRank
这个binSpect(二进制空间提取)也挺有意思:首先,对每个基因使用 kmeans 聚类(k = 2)或简单的秩阈值(默认值 = 30%)对表达值进行二分化(0、1)。接下来基于相邻细胞之间的二分化表达值计算列联表,并用作 Fisher 精确检验的输入以获得优势比估计值和 p 值。如果一个基因通常被发现在近端或邻近细胞中高度表达,那么它就被认为是一个空间基因。除了每个基因的优势比和 p 值之外,还计算了平均基因表达、高表达细胞的数量和中心细胞的数量(具有感兴趣基因的高表达并且具有该基因的多个高表达相邻细胞的细胞)。
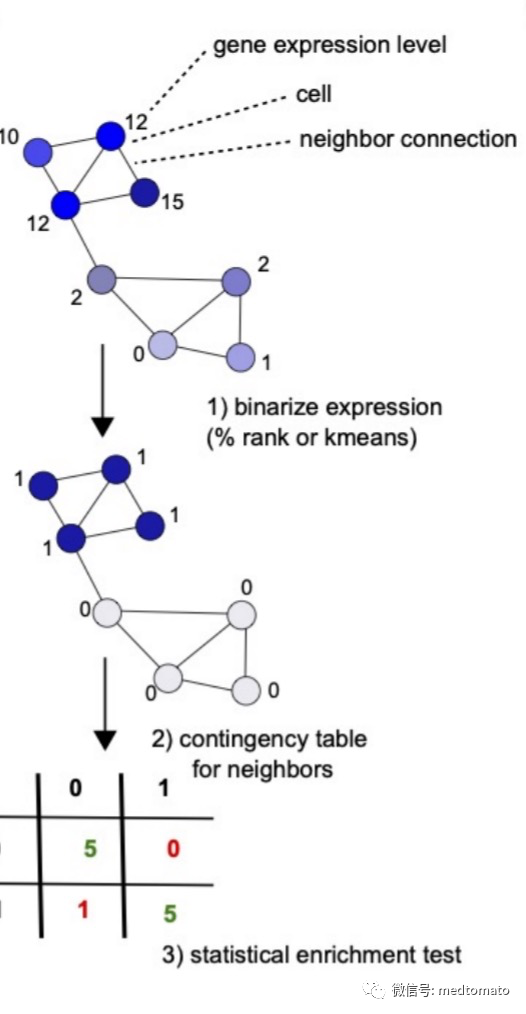
mini_visium = createSpatialNetwork(gobject = mini_visium,minimum_k = 2,maximum_distance_delaunay = 400)mini_visium = createSpatialNetwork(gobject = mini_visium,minimum_k = 2,method = 'kNN',k = 10)km_spatialgenes = binSpect(mini_visium)# This is the single parameter version of binSpect# 1. matrix binarization complete## 2. spatial enrichment test completed## 3. (optional) average expression of high expressing cells calculated## 4. (optional) number of high expressing cells calculated
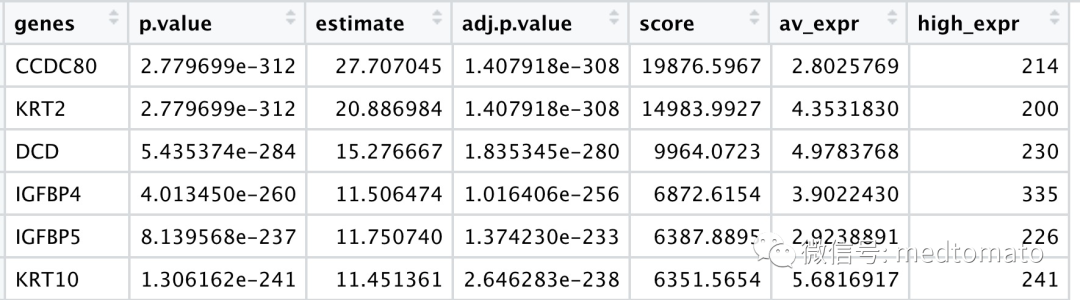
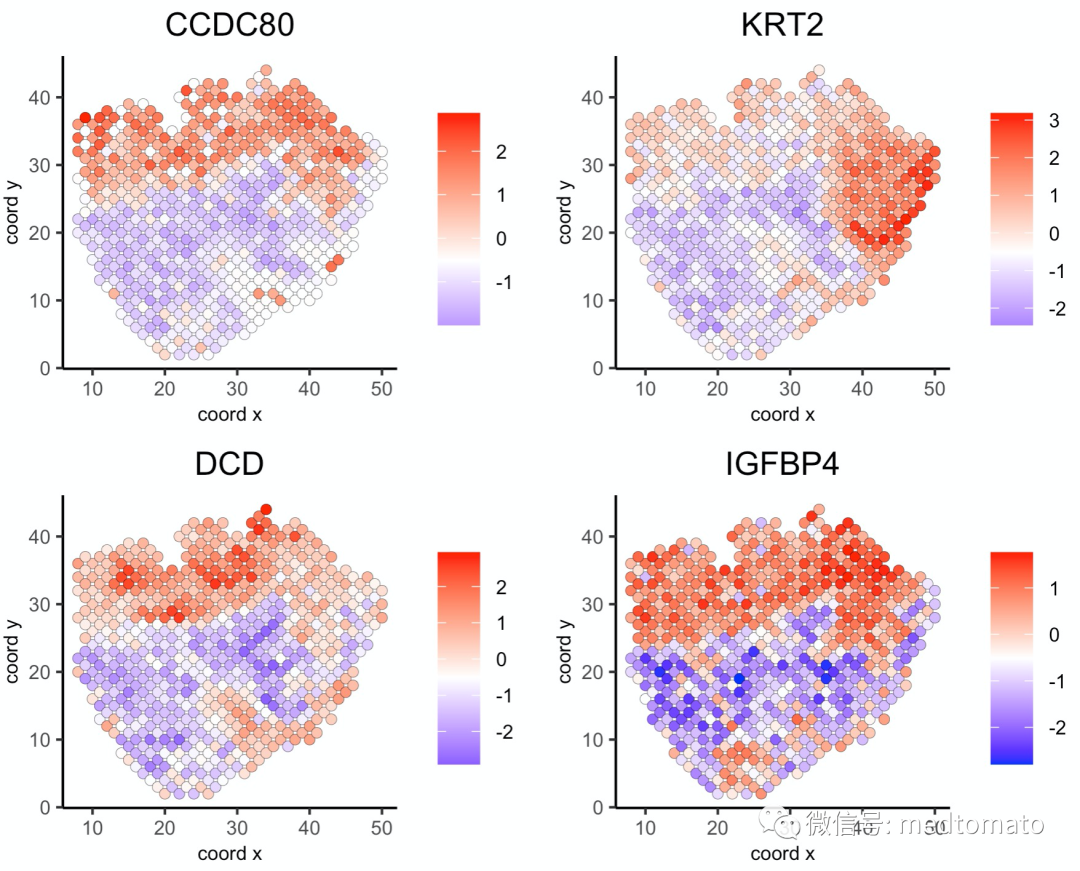
找出空间基因后可视化:
spatGenePlot(mini_visium, expression_values = 'scaled',genes = km_spatialgenes[1:4]$genes,point_shape = 'border',point_border_stroke = 0.1,show_network = F, network_color = 'lightgrey',point_size = 2,cow_n_col = 2)
# 1. calculate spatial correlation scoresext_spatial_genes = km_spatialgenes[1:1000]$genesspat_cor_netw_DT = detectSpatialCorGenes(mini_visium,method = 'network',spatial_network_name = 'Delaunay_network',subset_genes = ext_spatial_genes)# 2. cluster correlation scoresspat_cor_netw_DT = clusterSpatialCorGenes(spat_cor_netw_DT, name = 'spat_netw_clus', k = 8)heatmSpatialCorGenes(mini_visium, spatCorObject = spat_cor_netw_DT, use_clus_name = 'spat_netw_clus')netw_ranks = rankSpatialCorGroups(mini_visium, spatCorObject = spat_cor_netw_DT,use_clus_name = 'spat_netw_clus')top_netw_spat_cluster = showSpatialCorGenes(spat_cor_netw_DT, use_clus_name = 'spat_netw_clus',selected_clusters = 6,show_top_genes = 1)cluster_genes_DT = showSpatialCorGenes(spat_cor_netw_DT, use_clus_name = 'spat_netw_clus', show_top_genes = 1)cluster_genes = cluster_genes_DT$clus;names(cluster_genes) = cluster_genes_DT$gene_IDmini_visium = createMetagenes(mini_visium, gene_clusters = cluster_genes,name = 'cluster_metagene')
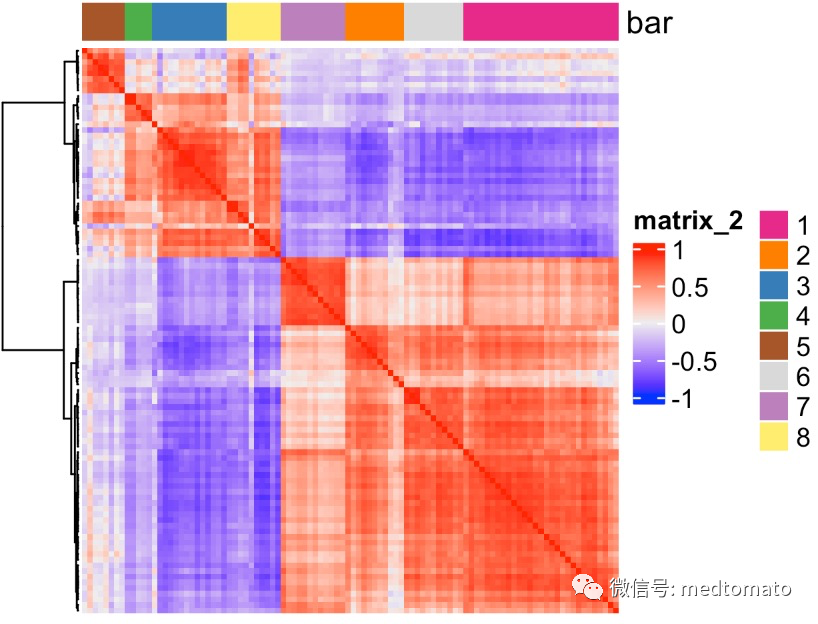
这里把空间基因分成了不同的pattern
netw_ranks = rankSpatialCorGroups(mini_visium, spatCorObject = spat_cor_netw_DT,use_clus_name = 'spat_netw_clus')cluster_genes_DT = showSpatialCorGenes(spat_cor_netw_DT,use_clus_name = 'spat_netw_clus',show_top_genes = 1)cluster_genes = cluster_genes_DT$clus;names(cluster_genes) = cluster_genes_DT$gene_ID
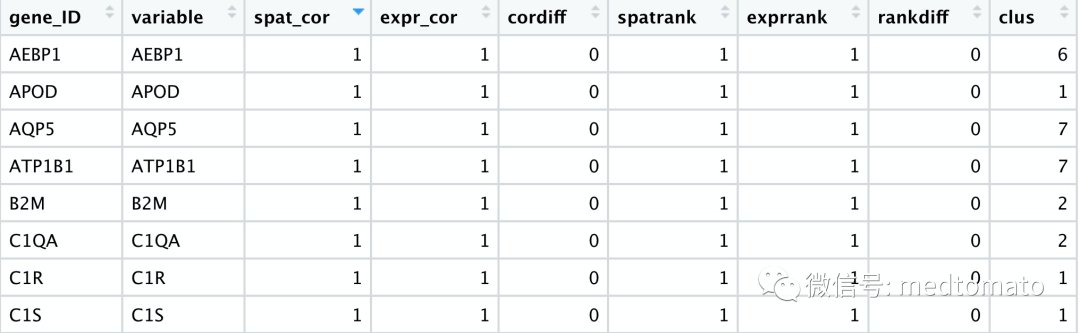
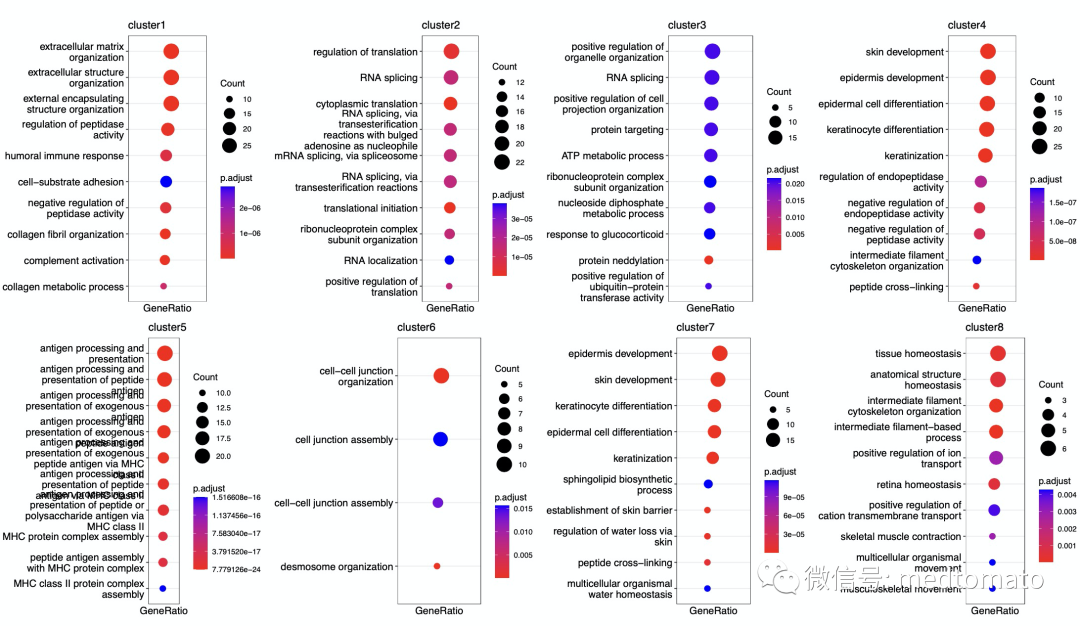
写到这里,我突然想到是否可以对这些pattern做个功能注释:
library(clusterProfiler)Sigbp <- lapply(Sig,enrichGO,OrgDb="org.Hs.eg.db",ont= "BP",keyType = "SYMBOL")bpplot <- lapply(names(Sigbp), function(z)dotplot(Sigbp[[z]], showCategory = 10) +ggtitle(z) + scale_x_discrete(labels=function(x) str_wrap(x, width=40)))cowplot::plot_grid(plotlist = bpplot, ncol = 4)
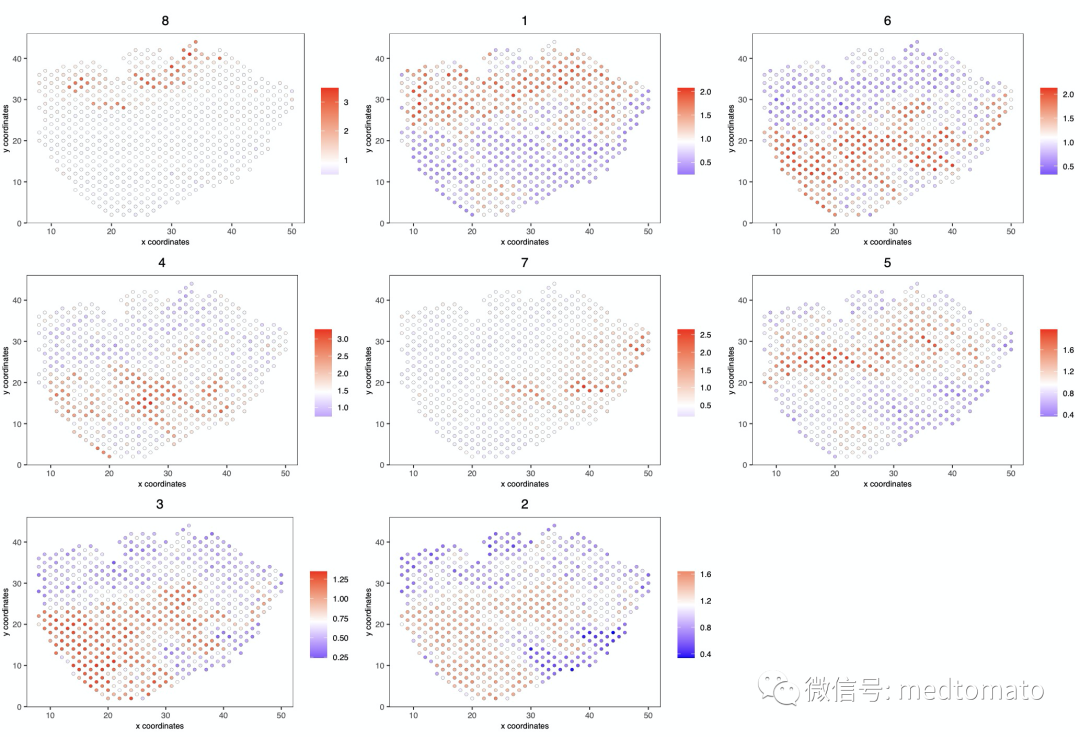
最后加上各个pattern的metagenes
# This function creates an average metagene for gene clusters.mini_visium = createMetagenes(mini_visium, gene_clusters = cluster_genes,name = 'cluster_metagene')spatCellPlot(mini_visium,spat_enr_names = 'cluster_metagene',cell_annotation_values = netw_ranks$clusters,point_size = 1.5, cow_n_col = 3)
Giotto的功能相当丰富啊,下次再更~

