




Hi-C数据分析相关的基本概念
染色质的层级结构
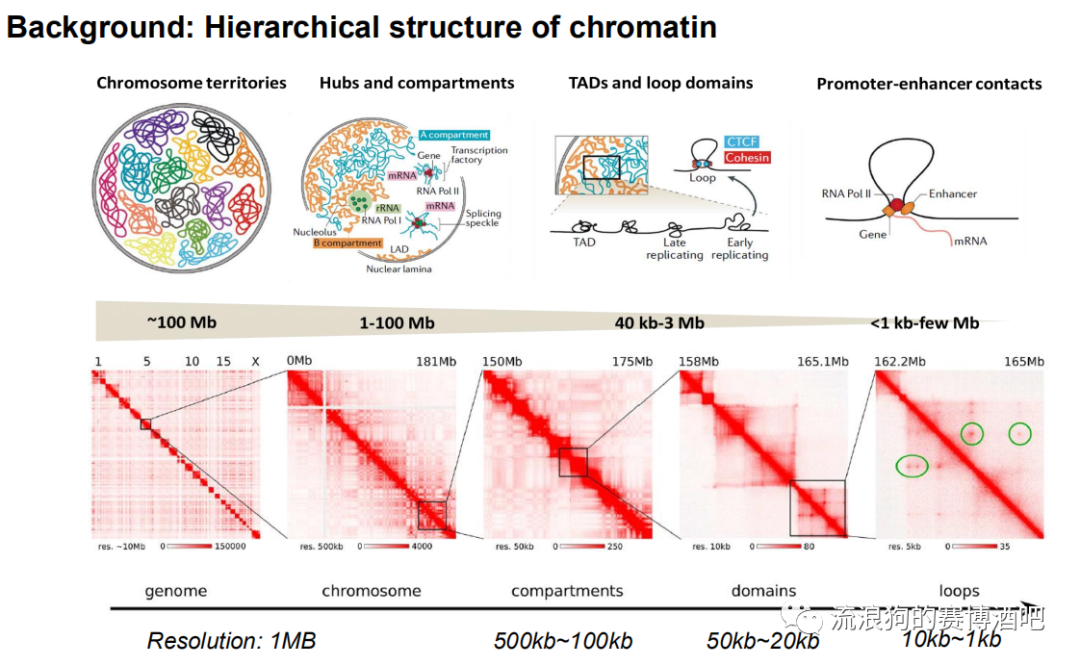 研究不同层级结构的互作关系需要不同的分辨率精度。上图示意了不同分辨率分别能反映的互作结构层次。关于互作结构层次概念,后文有详述。
研究不同层级结构的互作关系需要不同的分辨率精度。上图示意了不同分辨率分别能反映的互作结构层次。关于互作结构层次概念,后文有详述。
计数矩阵、热图及分辨率
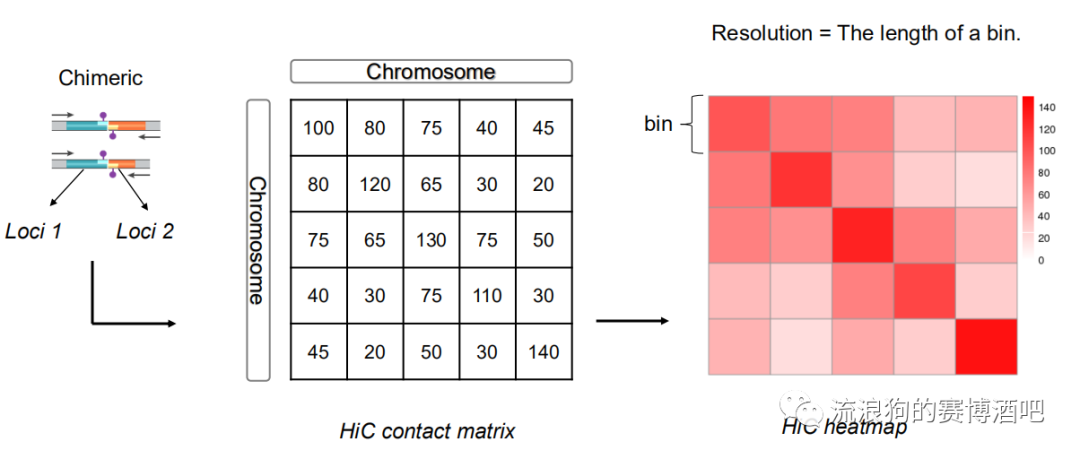 分辨率指的是进行互作关系统计时DNA被分割成的bin的长度。
分辨率指的是进行互作关系统计时DNA被分割成的bin的长度。
cis互作和trans互作
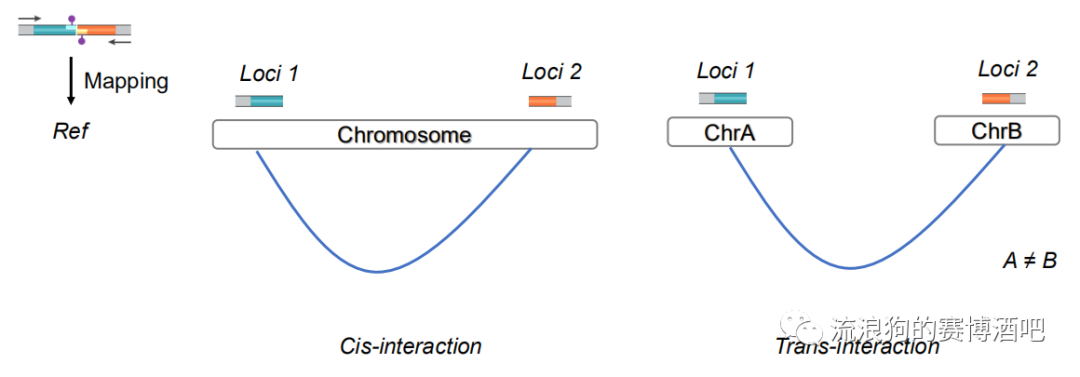 同一条染色质上的互作关系为cis,不同染色质上为trans
同一条染色质上的互作关系为cis,不同染色质上为trans
Hi-C分析流程
需要用到的软件概览

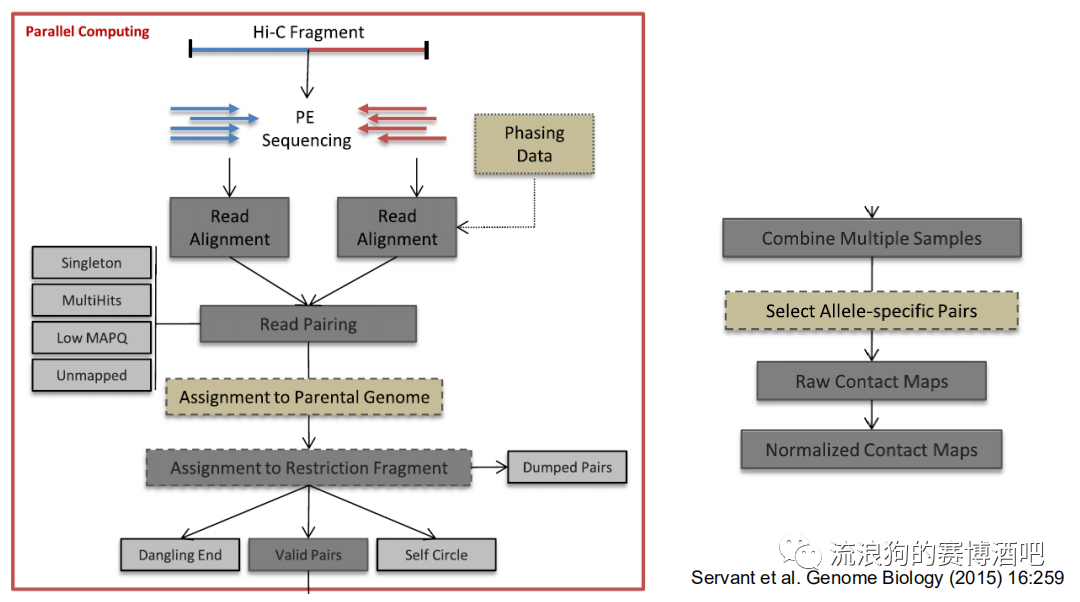
Hi-C pro
HiC-Pro workflow
HiC-Pro的安装:基于py2.7
在安装HiC-Pro之前需要先配置好如下环境
conda create --prefix=PATHWAY python=2.7
source activate PATHWAY
conda install -p PATHWAY -c bioconda bx-python
conda install -p PATHWAY -c ipyrad pysam
conda install -p PATHWAY -c conda-forge numpy
conda install -p PATHWAY -c conda-forge scipy
conda install -p PATHWAY -c bioconda iced复制
然后源码编译安装
tar -zxvf HiC-Pro-2.11.4.tar.gz
cd HiC-Pro-2.11.4
vi config-install.txt
make configure
make install复制
由于HiC-Pro是一个集成包,所以在初始化时需要定义一下调用的包的安装位置,如下图。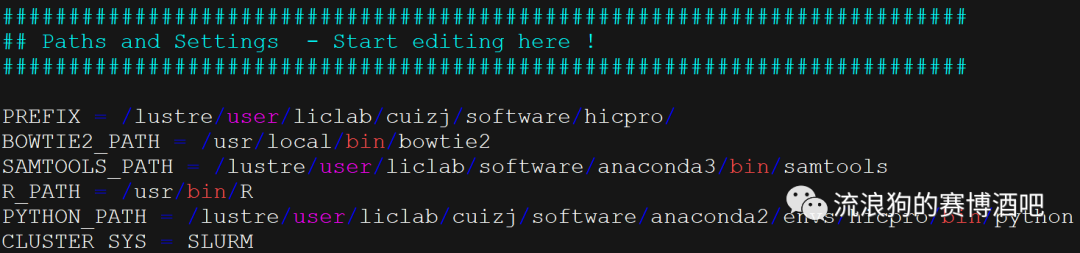
HiC-Pro的使用
https://github.com/nservant/HiC-Pro
Usage:
HiC-Pro -i INPUT -o OUTPUT -c CONFIG [-s ANALYSIS_STEP] [-p] [-h] [-v]
-i --input INPUT: input data folder; Must contains a folder per sample with input files
-o --output OUTPUT: output folder
-c --conf CONFIG: configuration file for Hi-C processing复制
-i:存放数据的文件夹,此文件夹下的每对双端测序文件都
需要重新建立一个文件夹。如下图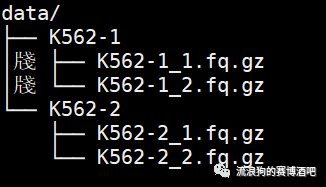
Config:
数据后缀名称的设置,需要与输入的fq.gz文件后缀一致
bowtie比对的参数设置。一般使用默认参数即可,仅需将BOWTIE2_IDX_PATH
设置为索引文件所在路径。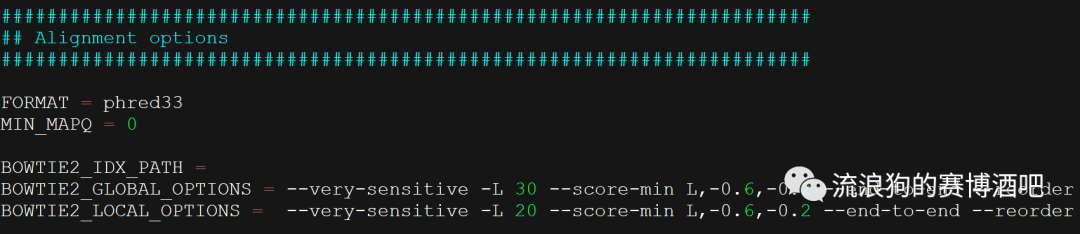
Annotation files
设置使用的参考基因组名称以及基因组大小。后者在软件安装时有专门的注释文件可供使用。Digestion Hi-C的GENOME_FRAGMENT
时酶切位点文件,同样为软件自备,只需选择合适的即可。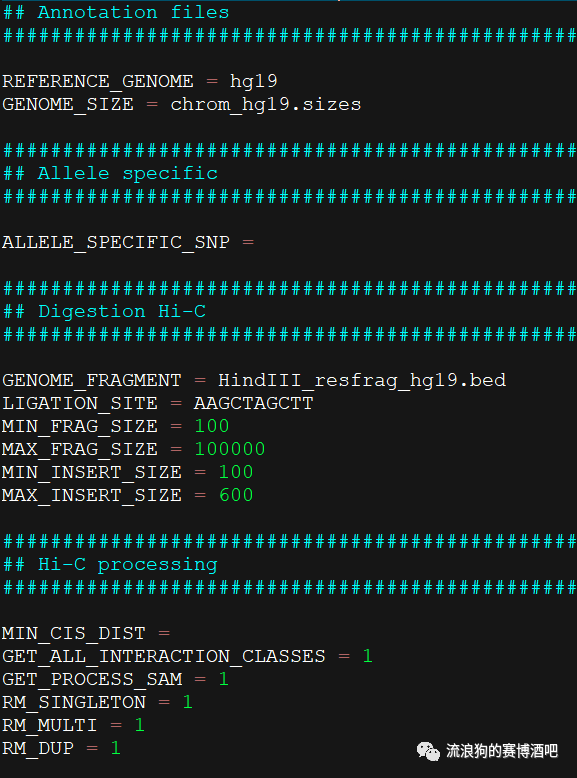
BIN_SIZE
就是矩阵分辨率,输入需要的数值即可。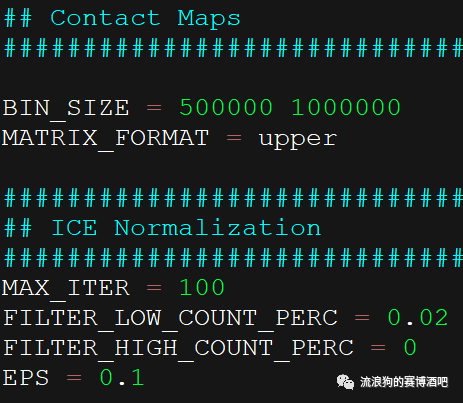
results
官方文档:http://nservant.github.io/HiC-Pro/RESULTS.html
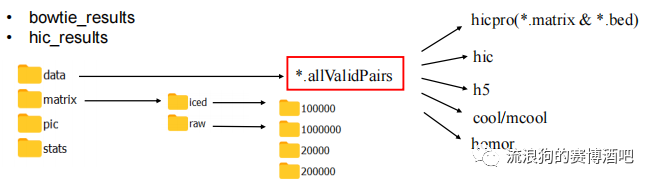 results分为bowtie比对结果和Hi-C结果
results分为bowtie比对结果和Hi-C结果
Hi-C结果中,最重要的就是*.allValidPairs
这个文件,后续数据分析都是基于这个文件。
matrix中的iced是校正后的数据,用的是ice方法。
*.allValidPairs一般包含12列信息:read name, chr_read1, pos_reads1, strand_read1, chr_read2, pos_reads2, strand_read2, fragment_size, res_frag_name1, res_frag_name2, mapq_read1, mapq_read2
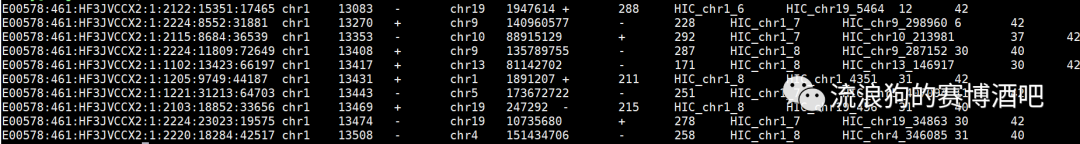
从*.allValidPairs可得到hicpro(*.matrix & *.bed)文件
.bed文件储存着bin的位置信息 .matrix储存互作count数量。
.matrix储存互作count数量。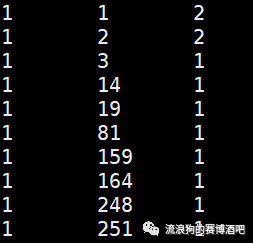
评估最高分辨率:juicer
https://github.com/aidenlab/juicer
判断标准:
某分辨率下,大于1000交互的bin所占的比例, 是否大于80%。
用法:
找到PATHWAY/juicer/juicer/misc/calculate_map_resolution.sh
脚本,运行即可
nohup sh calculate_map_resolution.sh *.allValidPairs coverage_output &复制
coverage_output
是自己指定的输出文件。运行结果:
Call A/B compartment
A/B compartment
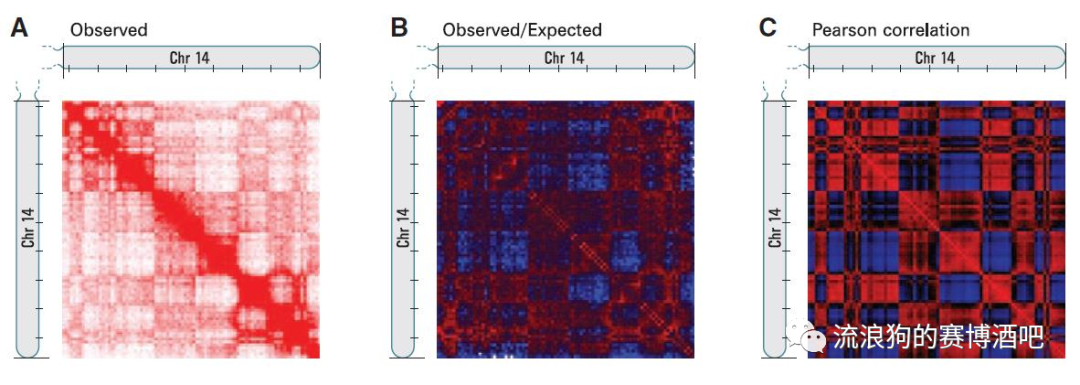 (Lieberman-Aiden et al., Science, 2010)
(Lieberman-Aiden et al., Science, 2010)
在这篇最早发表的文章中,作者发现把Hi-C结果标准化后,出现了区别非常明显的格子形状(c图)。将数据用PCA降维后,出现了AB两个主成分,一个是正的一个是负的,即A/B compartment。其中正的为A,负的为B。
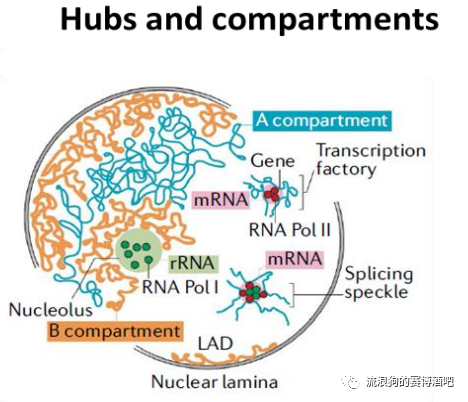
A compartments:开放的染色质,表达活跃,基因丰富,具有较高的GC含量,包含用于主动转录的组蛋白标记,通常位于细胞核的内部。
B compartments:关闭的染色质,表达不活跃,基因缺乏,结构紧凑,含有基因沉默的组蛋白标志物,位于核的外围
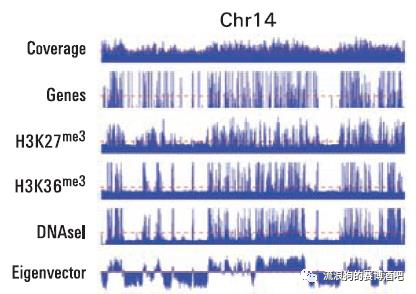 最后一行Eigenvecotr的正负区分了A/B compartments,结合上面几行图像信息可以明显看出这种特点
最后一行Eigenvecotr的正负区分了A/B compartments,结合上面几行图像信息可以明显看出这种特点
工具1:HiTC(pca.hic)

工具2:juicer (eigenvector)
https://github.com/aidenlab/juicer/wiki/Eigenvector
juicer只能单个染色体分析,所以要写循环。
for chr in {1..22}
do
java -jar juicer_tools.jar eigenvector <NONE/VC/VC_SQRT/KR> *.hic $chr BP <binsize>
./juicer_output/${chr}_eigen.txt
done复制
<NONE/VC/VC_SQRT/KR>:选择标准化方式。关于这几种标准化方式的特点,在github上查到了答案但是看不懂,先放这吧。不知有无大佬能解答:
VC: vanilla coverage (overcorrects low coverage loci)
VC_SQRT: square root vanilla coverage (creates a matrix whose row and column sums are all approximately equal)
KR : Knight and Ruiz normalization (works at both low and high resolutions.)
Ref: Knight, P., and Ruiz, D. (2012). A fast algorithm for matrix balancing. IMA J. Numer. Anal. Published online October 26, 2012. http://dx.doi.org/10.1093/imanum/drs019.
https://github.com/aidenlab/juicer/issues/19
< binsize> :选择分辨率大小
*.hic
到*.allValidPairs
的格式转换:
*.hic
是需要输入的数据文件。而将数据格式从*.allValidPairs
转换为*.hic
需要用到HiC-Pro的一个脚本:
PATHWAY/HiC-Pro_2.11.4/bin/utils/hicpro2juicebox.sh -i ./*.allValidPairs -g PATHWAY/HiCPro_2.11.4/annotation/chrom_hg19.sizes -j PATHWAY/juicer/scripts/CPU/common/juicer_tools.jar
-t tmp -o ./复制
命令里除了*.allValidPairs
文件位置,还需要给出基因组注释大小文件的位置以及juicer的安装位置。
Call TADs
TADs 和 loop
TADs:topological associated domains 拓扑相关结构域。是指在染色质区室中,互相作用相对频繁的基因组区域。(第一行的每一个红色三角就是一个TAD)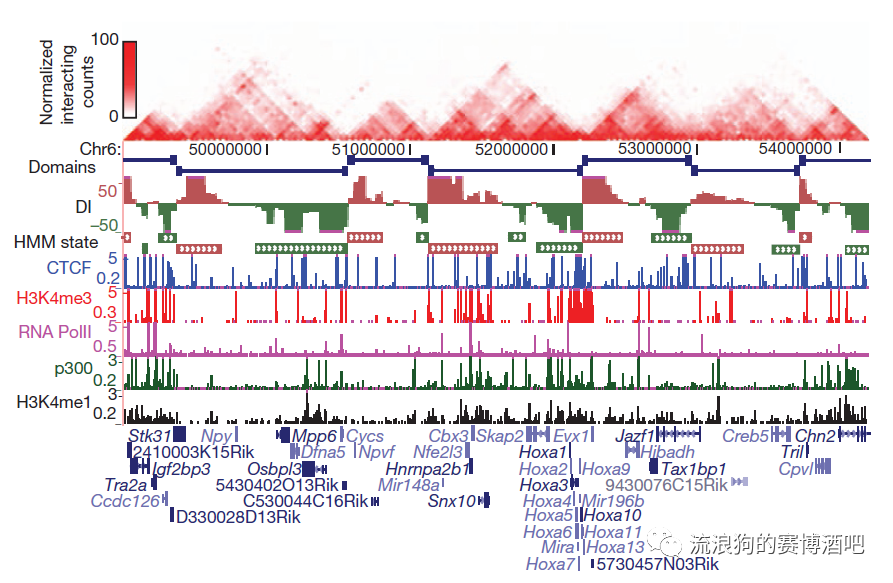
在哺乳动物基因组中,TAD通常由CTCF这个转录抑制因子给分割开来。CTCF还会和Cohesin蛋白复合物结合,帮助基因组形成相对稳定的三维结构。
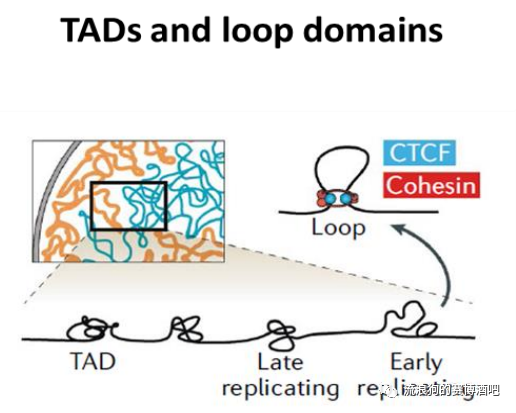
正由于此,两个TAD之间的转录活性非常低(转录需要打开DNA),而结合CTCF等转录抑制因子的DNA元件,也被称为insulator(绝缘子)。
在TAD内部转录非常活跃。CTCF在帮助基因组DNA凹造型的同时,把DNA元件给绑到了一起。而这样相互作用的元件,通常是enhancer(增强子)和promoter(启动子),他们往往分布在相距很远的染色质区域,却因为CTCF蛋白在三维空间中聚集在一起,我们把这种结构称之为loop。
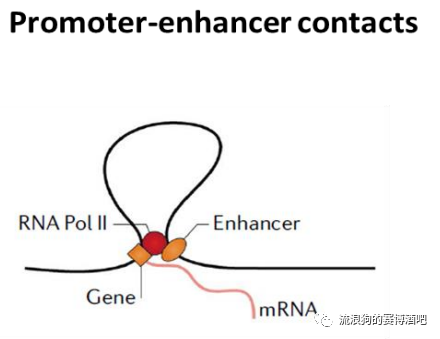
(部分解释引自知乎https://www.zhihu.com/question/50270622/answer/1715108352)
Call TADs
由于TAD结构对功能有非常大的影响,因此识别TAD边界至关重要。
现有有如下几种方法:
Directionality Index,DI
Insulation Score
Arrowhead
TADbit
DomainCaller
TADtree
其中前三种较为常用。
Call TADs: DI
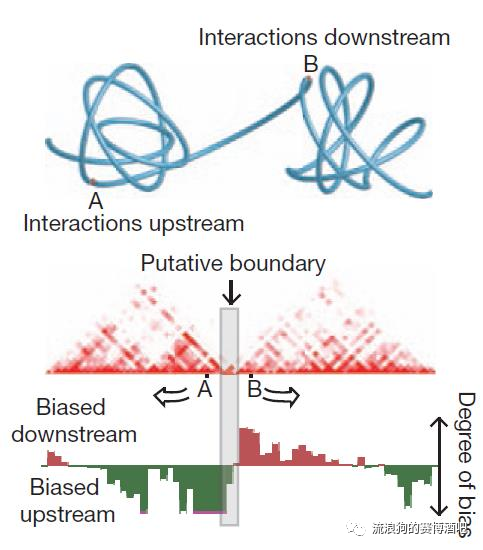 对于TAD边界位置来说,它的左右位置互作方向是相反的。如上图,A点偏好上游,而B点偏好下游。directionality index(方向指数)这个方法就是根据这个原理设计的。
对于TAD边界位置来说,它的左右位置互作方向是相反的。如上图,A点偏好上游,而B点偏好下游。directionality index(方向指数)这个方法就是根据这个原理设计的。
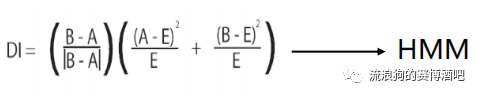
DI-HMM 方法首先采用方向指数(directionality index)来表征一个染色体区域与上下游交互作用的偏差,当这个偏差出现符号跳转时,意味着可能出现 TAD 的边界.在方向指数中利用隐马尔可夫模型可以推断出 TAD 的具体位置。(但是HMM模型到底是如何在算法中起作用的?)
Call TADs: Insulation Score
https://github.com/dekkerlab/crane-nature-2015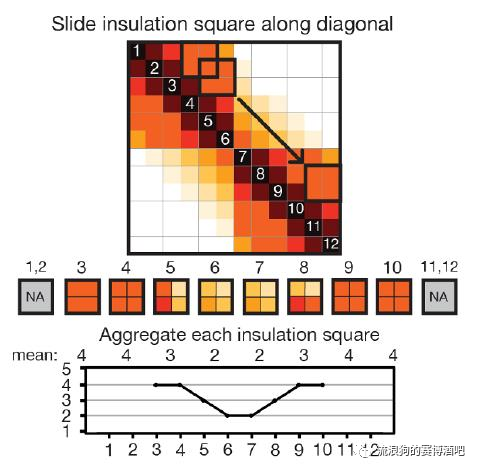 首先在热图上定义一个滑动窗口,在滑动过程中统计每个窗口中互作的数量,这样可以得到下面那条线。这样就可以得到互作数量发生转变的位置。
首先在热图上定义一个滑动窗口,在滑动过程中统计每个窗口中互作的数量,这样可以得到下面那条线。这样就可以得到互作数量发生转变的位置。
perl matrix2insulation.pl -i *.dense_header.matrix -is 500000 -ids 250000 -im mean -nt 0.1 -o ./复制
参数解释
i: 交互矩阵
is: insulation square,默认500000,动物推荐500000,植物推荐200000-250000.
ids: insulation delta span,默认250000,植物推荐100000-200000. is必须要比ids大.
nt: delta bound阈值,提高阈值可以过滤一些边界不明显的TAD.
bmoe: boundary margin of error.一般不予设定。
如何得到交互矩阵
hicpro(*.matrix & *.bed) → IS input matrix
Step1: 将稀疏矩阵转成稠密矩阵。
(稀疏矩阵:矩阵中0元素的个数远大于非零,且0元素分布无规律;稠密矩阵反之)
Python2.7 PATHWAY/HiC-Pro_2.11.4/bin/utils/sparseToDense.py *_50k_iced.matrix -b *_50k_abs.bed --perchr > mESC_50k.log 2>&1复制
Step2: 添加表头
采用R或者python/perl
表头形式bin6000000|ce10|chrX:1-10001
bin的顺序|物种参考基因组的名字|染色体位置信息
下图是利用R添加表头的代码示例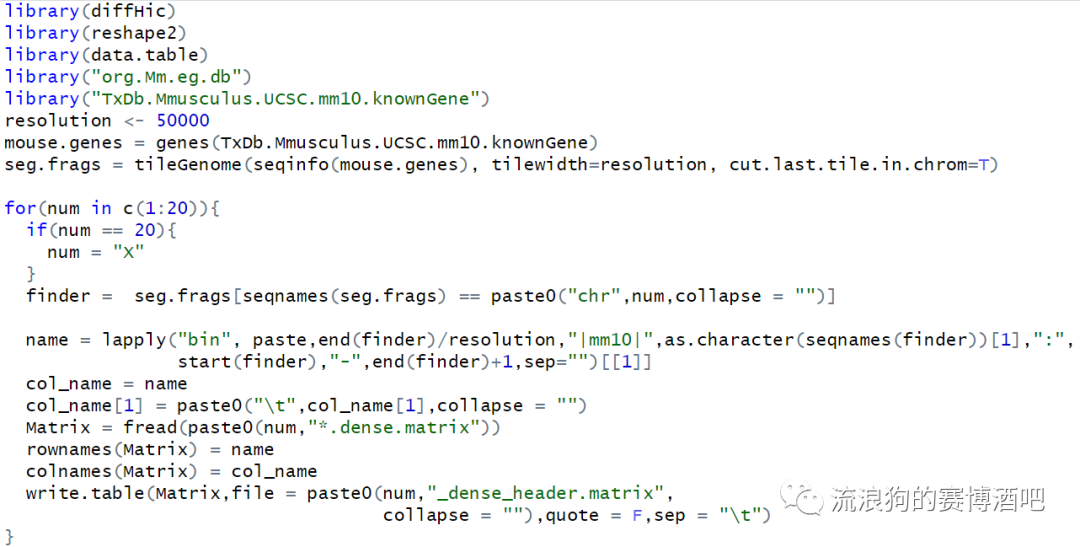 注意:
注意:
line5调用的参考基因组,之前用的哪个版本比对,这时候就调用哪个版本。
line6要输入数据分析所使用的分辨率数值
输出结果
输出4个文件
.insulation.bedGraph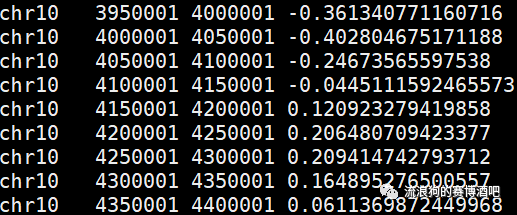 前三列是每个BIN的位置信息,最后一列是insulation score的得分。
前三列是每个BIN的位置信息,最后一列是insulation score的得分。
.insulation.boundaries.bed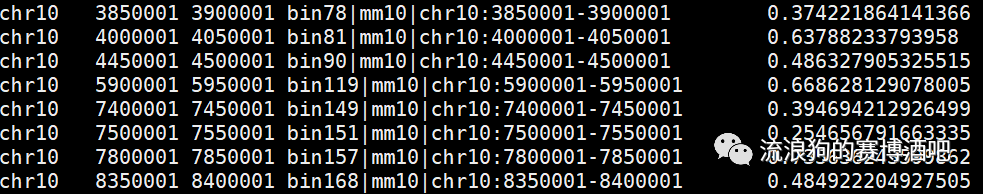 边界信息文件。第四列是包含位置信息的表头,最后一列是边界强度的得分情况
边界信息文件。第四列是包含位置信息的表头,最后一列是边界强度的得分情况
Call TADs: Arrowhead
https://github.com/aidenlab/juicer/wiki/Arrowhead
是一个juicer工具。
java -Xmx8g -jar PATHWAY/juicer/scripts/CPU/common/juicer_tools.jar arrowhead ./*.hic --
threads 8 -r 10000 -k KR ./contact_domains_10kb复制
Call loops: HiCCUPS
https://github.com/aidenlab/juicer/wiki/HiCCUPS
同样是juicer工具。可以用GPU或CPU运算,一般还是选择CPU。但是GPU运算速度快一点。
java -Xmx8g -jar PATHWAY/juicer/scripts/CPU/common/juicer_tools.jar hiccups --cpu
--threads 8 -r 5000 -k KR ./*.hic ./复制
可视化工具
有以下几种:
• Juicebox
• WashU: http://epigenomegateway.wustl.edu/
• HiTC
• HiCPlotter
• HiCExplorer
• R
这个比较直观,就不记了。用到的时候再说吧。
评论

 0 点赞 0 点赞
0 点赞 0 点赞